 Distributed computing simulations in molecular dynamics area are very popular and they are frequently used in modern chemistry, pharmacology, pharmaceutical chemistry and biochemistry. Computing models that can simulate and predict molecule interactions with well-known proteins give enormous contribution in resolving complex problems. These simulations can reduce the costs of developing new cures, which takes 12-15 years and costs between 600 and 800 million US dollars, and accelerate process of compounds selection that are considered as part of new medicine. Mixture of molecular dynamics and distributed computing is very relevant in modern research of potential cures.
Distributed computing simulations in molecular dynamics area are very popular and they are frequently used in modern chemistry, pharmacology, pharmaceutical chemistry and biochemistry. Computing models that can simulate and predict molecule interactions with well-known proteins give enormous contribution in resolving complex problems. These simulations can reduce the costs of developing new cures, which takes 12-15 years and costs between 600 and 800 million US dollars, and accelerate process of compounds selection that are considered as part of new medicine. Mixture of molecular dynamics and distributed computing is very relevant in modern research of potential cures.
Molecular dynamics simulations are focused on the examination of interactions between synthetized molecule and proteins of the cell membrane. The simulations are based on GROMACS (GROningenMAchine for Chemical Simulations) and NAMD (NAnoscale Molecular Dynamics), applications for parallel simulations in the molecular dynamics. Simulations are performed with intention to show possibility of creation good models for further analysis. Using this molecular models it should be possible to study the interaction of organic molecules, which previously were not synthesized in the laboratory (which significantly reduces the time and research, but also the price of the process), with appropriate protein (i.e. receptor). Additional data will be available about possible further structural units of the molecule, which would be responsible for additional or maybe even stronger interactions with receptors (proteins) of cell membrane. In that manner, the same pharmacological properties of the test molecule would be improved. Completely computationally intensive simulation process is accelerated by using dozens or hundreds of computing cores, which gives a significant advantage of using computer models in comparison to the traditional way of synthesis of new molecules, and subsequent analysis of their biological activity. Parallel simulations were performed on FINKI HPC, which is part of VI SEEM eInfrastructure. By using parallel data processing, simulations are noticeably accelerated, with efficiency more than 92%. The results were received quicker, providing the opportunity to try many combinations before recommendations for reaction testing in the laboratory. The obtained dataset, workflow and molecule trajectories were uploaded on VI-SEEM repo and VRE portal for further analysis.
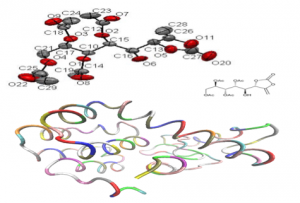
17 new and previously unknown molecules were synthetized, which are based on the existence of enol carbonate structural units. Specified unit is a common structural motive of many natural products and physiologically active compounds, which makes molecules suitable and interesting for further analysis. One of synthetized compounds was (2R, 3R, 4R, 5S)-5-hydroxy-5-((S)-5-methylene-2-oxo-1, 3-dioxolan-4-yl) pentane-1, 2, 3, 4- tetraacetate and which was a basis for further research. After structural analysis and determination of precise layout of all the atoms in space, molecule was converted into standard chemical format, visualized and prepared for computer simulations.
Links:
- https://repo.vi-seem.eu/handle/21.15102/VISEEM-76
- https://vre.vi-seem.eu/workflow-pipeline-and-software-tools-repository/scientific-workflows
Published Papers :
– M. Bigović, Ž. Zečević, L. Filipović, B. Krstajić, “Komparacija računarskog modela molekula sa eksperimentalnim rezultatima”, Information technology – IT’17, Žabljak, 2017, ISBN:978-86-85775-20-8
– M. Bigović, Ž. Zečević, L. Filipović, B. Krstajić, “Verification of the three-dimensional structure of synthesized molecule by molecular dynamicsimulations”, IEEE EUROCON 2017, Ohrid, FYRo Macedonia