Issues related to drug delivery are of essential importance to contemporary molecular pharmaceutics. Molecular modeling approach is of substantial importance in the course of efficient design and development of conventional dosage forms as well as novel micro/nano carrier systemsforactive pharmaceutical ingredients – API(from in-silico to industrial level scaling). Namely, during the preformulation studies proper characterization of API, selected excipients/polymer sand their interactions are essential for efficient development of the new drug product. Therefore,in our application, we develop and implement new, robust computational modeling approaches to study the structural and vibrational spectroscopic properties of different API`s as well as to investigate the physics and chemistry of interactions between drug molecules and their potential carriers, under realistic conditions encountered in biological systems. The intermolecular interactions relevant to the present application span a rather wide range, in both qualitative and quantitative sense. The methodologies that we develop are inherently hybrid. They are based on sequential application of statistical physics simulations and quantum mechanical calculations to the systems of interest. Statistical physics phase is, in our approach, mostly based on ab initio molecular dynamics (MD) simulations, in the framework of atom-centered density matrix propagation scheme (ADMP) or Born-Oppenheimer MD. In certain cases, we also implement classical MD and Monte Carlo. Quantum mechanical phase involves exact computation of molecular properties relevant to drug – nano-carrier intermolecular interactions. Final judgment concerning the usefulness and applicability of the developed models is brought up by comparison with relevant experimental data. At the current stage of development, we have prepared workflows for most of the specific computational purposes andwe have also written most of the codes for data analysis. In parallel, we havealso carried out numerous tests along with some productive computations.
To implement our application, we have been given excellent opportunity to use the Supercomputing Facility at Bibliotheca Alexandrina (BA) in Cairo, Egypt. The supercomputing facility at BA is actually a HPC cluster with peak performance of 11.8 TFLOPS, composed of 130 computing nodes, each with two Intel Quadcore Xeon 2.83 GHz CPUs, a 36-TByte storage system, dual port infiniband (10 Gbps) and a GigaEthernet network port.
We have already made workflows for some particular computational purposes, along with certain outputs and datasetsavailable to the community and the other partners of the VI-SEEM project (see the repositories site).We have also provided links to the scientific publications that have arisenup to this point from our application.
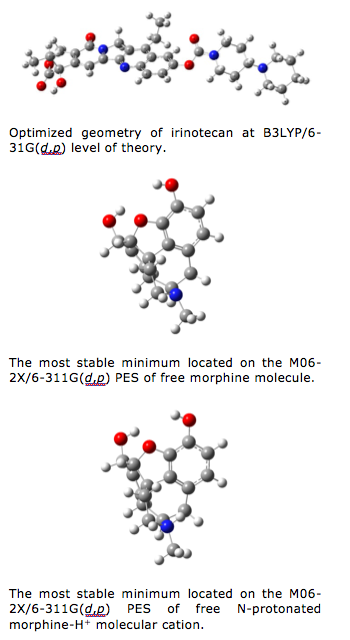
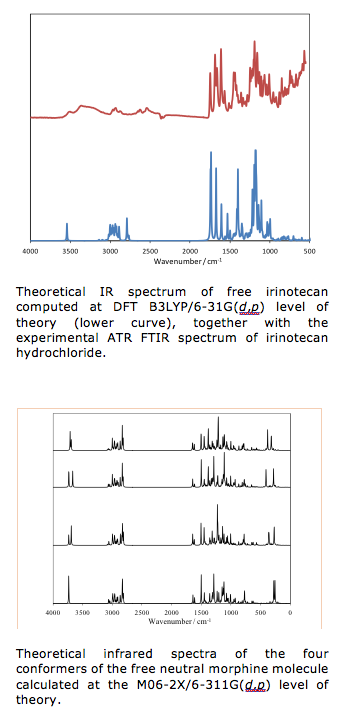
Up to now, we have successfully implemented our hybrid computational approach to study the structural and vibrational spectroscopic properties of hydrophilic drug irinotecane. This actually served as a first step towards a multiscale model that would lead us to an in-depth understanding of the physics and chemistry behind the processes of incorporation of this molecular system into nanoparticles built up by poly lactic-co-clycolic acid copolymer and co-adsorbed polyethylene oxide – polypropylene – polyethylene oxide. Comparison of our theoretical results with the experimental ATR FT IR data demonstrated the validity of the approach. Further, we have also studied the vibrational spectroscopic properties of morphine as well as protonated morphine-H+ cationic species and compared our theoretical predictions with the experimental FT IR data. Not only that our theoretical approach has enabled solid support to the empirical assignment of the spectral bands, but it also implied certain fundamental insights into the structural properties of the solid-state form of this exceptionally important compound. Aside for this, the current work serves as a starting point for further studies of the spectroscopic manifestations of morphine incorporation into prolonged release oral formulations, where different polymers or preparation methods are used in order to obtain suitable morphine release. In this context, we have also theoretically modeled the kinetics of the process of morphine in vitro release from developed prolonged release formulations.